Scheme 6:
The purpose of this paper is to present a unifying, qualitative framework for acid/base concepts that students may apply to a variety of situations in aqueous solution, the medium to which application of acid-base concepts is usually restricted in introductory treatments. The essential idea of the framework is that BL acidity in aqueous solution is a consequence of the interaction of cations with water to an extent governed by their inherent Lewis acidities. Within the framework, the similarity of seemingly dissimilar compounds is stressed. Thus, for example, substances such as NaOH, CH3COOH, and H2SO4, ordinarily viewed as very different in nature, may all be thought of as arising from the hydrolysis of water molecules promoted by their interaction with the appropriate cation (Na+, CH3C3+, and S6+, respectively). I will show that the phenomenon of cation hydrolysis, with its origins in the Lewis acidity of the cation, is the foundation for proton transfer (BL) processes in aqueous solution. As such it deserves much more than the very limited treatment usually given it in introductory texts (3). I hope that my perspectives on and enhancements of ideas proposed by others (4-6) will make a broad view of acid-base concepts more accessible to students at the introductory level.
The Relationship of Bronsted-Lowry to Lewis Theory.
The focus in BL theory is on the proton, H+. The fundamental process considered
in the theory is the proton transfer, 1:
HB + B'---> (HB')+ + B- (1)
HB and (HB')+ are BL acids (proton donors)
because they donate protons to the BL bases, B' and B- respectively.
In Lewis theory, the focus is on the electron pair. The fundamental process
considered in the theory is adduct formation, 2. The reverse process is
adduct dissociation.
A + :B ---> AB (2)
In this process, B is a base because it donates an electron
pair to the acid, A. The result is the adduct (addition product), AB. Of
the processes 1 and 2, 2 is simpler because it involves bond formation
only. Process 1 involves bond breaking (in HB) followed by bond formation
to form (HB')+ and may be thought of as occuring in two steps:
HB ---> H+ + :B- (1a)
H+ + :B'---> (HB')+ (1b)
Process 1a is dissociation of the Lewis adduct, HB; process 1b is formation of the Lewis adduct, (HB')+. This very brief overview of the two theories leads to the following generalizations:
The Bronsted-Lowry Spectrum of Water. The complete
series of BL acid-base reactions of water can be expressed in terms of
the four species given below:
H3O+, H2O, OH-,
O2- (3)
<--- increasing acidity
increasing basicity --->
Acidity and basicity vary along the series as indicated.
The species, H3O+, is the strongest acid that can
exist in water. Although there is still a nonbonding electron pair on oxygen,
the positive charge of H3O+ makes addition of a second
proton unlikely; H3O+ ordinarily exhibits no Bronsted
basicity. H2O functions as both an acid and a base, depending
on circumstances; and OH- usually functions as a base. However,
it has one remaining proton that can conceivably be removed, and can be
viewed as a potential BL acid (proton donor) with conjugate base O2-.
The oxide ion exists in solid oxides of a number of metals (e.g., Na2O,
MgO, Al2O3). However, it is too basic to persist
in water; instead, it accepts a proton via reaction 4:
O2- + H2O ---> 2OH-
(4)
The Lewis Acidity of Cations. Now we focus on the
factors that determine the inherent Lewis acidity of cations. There are
two factors of major importance: the charge, z+, and the size (radius,
r) of the cation (4,6,14). In addition to these, the electronegativity
is of considerably less dramatic importance. Thus Lewis acidity can be
quantitatively treated in terms of the quantity Z2/r (4), as
shown in equation 5:
Cation acidity = kZ2/r (5)
The more positive the cation charge, the greater the tendency of the cation to accept electron pairs from Lewis bases. For cations of a given charge level (say 3+) acidity increases with decreasing size. In general, cations may be expected to form adducts with Lewis bases in an attempt to partially or completely neutralize their inherent acidity. Strong acids, with high positive charge and/or small radius, must form adducts either with very strong bases (e.g., O2-) or with a larger number of weaker bases (e.g., OH- or H2O) in order that the resulting adduct be neutral. In contrast, the acidity of weak Lewis acids, with low positive charge and/or large radius, is readily offset by fewer and/or weaker bases.
Cations in Water. I now consider the fate of a
cation, Mz+, when introduced to the solvent water. Here I intend
to include not only "real" cations such as Na+, Ca2+,
and Al3+, but also hypothetical cations, for example, N5+,
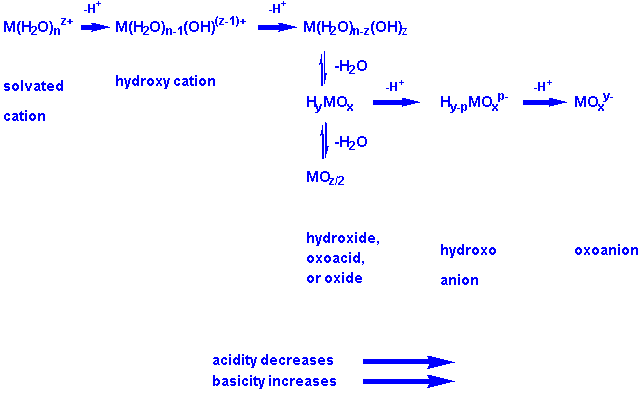
Se6+, and Br7+. Scheme 6 shows the sequence of reactions
through which a cation may be imagined to proceed in aqueous solution.
Scheme 6:
The inherent Lewis acidity of the cation, Mz+, determines how far along the sequence of species it proceeds. The more acidic the cation--that is, the more tendency it has to accept electron pairs from molecules (H2O) or ions (OH-, O2-) attached to it--the more it polarizes bound water molecules, and the more protons it loses. With this essential idea in hand, I will discuss the scheme in step-by-step detail.
Species A. When dissolved in water, the Lewis-acidic cation attracts the negative ends of the dipoles of a number of water molecules, and bonds to them via a Lewis acid base interaction. A lone pair of the oxygen atom of water is donated to the cation in the process. Species A is the resulting aquo adduct, in which the cation is bound to n water molecules. The value of n is frequently six. A is called a complex ion, and is a potential BL acid. Most cations of 1+ charge (e.g., Na+) are inherently weak Lewis acids, and form complexes that do not proceed along the sequence past point A. Thus complexes such as Na(H2O)6+ do not function as proton donors (BL acids) in aqueous solution to an appreciable extent.
Process 6a, written in full below, is a proton transfer
process, no different in principal from the reaction of, say, acetic acid
with water.
M(H2O)nz+(A)
+ H2O ---> M(H2O)n-1(OH)(z-1)+(B)
+ H3O+ (6a)
In 6a, the complex ion, A, donates a proton from a bound water molecule to a molecule of solvent water. This process is facilitated by the positive charge of the cation, Mz+, which drains electron density from the O atoms attached to it. These in turn drain density from the protons bound to them. A proton transfers to a solvent molecule, from which it receives more electron density. This sequence of electron drainage--induction--is illustrated in Figure 2.
Species A behaves as a BL acid (proton donor) as a result of the inherent Lewis acidity (electron pair acceptor ability) of the cation, Mz+. Thus the BL acidity of A is a consequence of Lewis acidity of the central cation of A.
Species B. For z > 2, this species is called a hydroxo cation. B is a weaker BL acid than A because the hydroxide ion more effectively neutralizes the inherent Lewis acidity of the cation than did the water molecule from which it was generated. Most cations with 2+ charge (e.g., Mg2+), stop at this stage of the sequence.
Process 6b is shown in full below:
M(H2O)n-1(OH)(z-1)+(B)
+ (z-1)H2O --->M(OH)z(H2O)n-z(C)
+ (z-1)H3O+ (6b)
If the aquo complex A is sufficiently acidic, it may lose more than one proton. Process 6b shows the loss of sufficient H+ ions to generate C, the neutral hydroxide. Many cations of 3+ charge (for example, Al3+) proceed to this point in the sequence. The Lewis acidity of a 3+ cation is sufficient to cause the aquo complex, A, to function as a triprotic BL acid.
Process 6c is written in full below:
M(OH)z(H2O)n-z(C)
---> HyMOx(D) + (n-x)H2O (6c)
This is not a proton transfer process. Instead it involves loss of water by the neutral hydroxide to form species D, an oxoacid. The common oxoacids may thus be considered to have their geneses in the inherent Lewis acidity of their central cations. Process 6c occurs if crowding of water and hydroxide species around Mz+ is severe in species C. The effect is to reduce the number of oxygen atoms attached to M, relieving steric strain. Only very small, highly charged cations progress along the series to point D. An example is the cation, P5+, which may be viewed as originating in a compound like PF5 or PCl5. It is just these species of high charge that tend to undergo process 6c, because the number of hydroxide anions required to produce the neutral hydroxide exceeds a viable coordination number for the cation. Typically elements of periods 3 and 4 are limited to a coordination number of four oxo or hydroxo ligands. To achieve this, the neutral hydroxide, P(OH)5, in which the coordination number of phosphorus is five, loses one water molecule to produce the oxoacid, PO(OH)3, with an acceptable coordination number for the central ion. In like fashion, the neutral hydroxides S(OH)6 and Cl(OH)7 far exceed acceptable coordination numbers for S6+ and Cl7+. They lose, respectively, two and three molecules of water to produce the familiar oxoacids, SO2(OH)2 and ClO3(OH). A procedure for predicting the number of water molecules involved in conversion of C to D has been published (15). I emphasize that for strongly acidic cations such as P5+, S6+, and Cl7+, none of the species A, B, or C is actually observable in aqueous solution. However, it is the premise of this paper that it is useful to think of them as (hypothetical) stages in the formation of the known oxoacids from the "cations" in halides. Such a mental framework enables a unified conceptualization of the oxo-, oxo-hydroxo, and hydroxo species of the elements that is not otherwise apparent.
Process 6d is again a proton transfer, this time from
the oxoacid to a molecule of water.
HyMOx(D) + H2O
---> H3O+ + H(y-1)MOx-(E)
(6d)
A cation that proceeds to this stage is S6+. It forms the oxoacid, H2SO4, which transfers a proton to H2O to give HSO4-. E is called a hydroxoanion, because it is anionic and contains at least one OH- bound to M. The BL acidity of oxoacids, HyMOx, is due to residual Lewis acidity of Mz+ that is incompletely neutralized by the oxo and hydroxo ligands of the oxoacid.
Process 6e is the final proton transfer that can occur.
Very highly charged, small cations such as Cl7+ and N5+
proceed all the way to species F, the oxoanion:
H(y-1)MOx-(E)
+ H2O ---> H3O+ + MOxy-(F)
(6e)
Generalizations from Sequence 6. Having described the sequence of processes possible for a cation following its dissolution in water, I would like to relate progression of a cation along sequence 6 to the Bronsted spectrum of water, in which basicity increases markedly from H2O to O2- (the oxide ion). The following generalizations can be made:
| Z2/r | Form at pH 7 | Example |
|---|---|---|
| 0-0.04 | hydrated cation | Na(H2O)6+ |
| 0.04-0.22 | hydroxide, oxide,
or oxide-hydroxide |
Al(H2O)3(OH)3 |
| 0.22-0.8 | oxide-hydroxide or
hydroxo anion |
SeO3(OH)- |
| > 0.8 | oxoanion | BrO4- |
These examples illustrate the use of the Lewis concepts in rationalizing the Bronsted acidity of species resulting from cation hydrolysis in aqueous solution. In terms of the framework presented in this paper, species such as Na(H2O)6+, Al(OH)3, and H2SO4 are apparently closely related. They are all Lewis adducts, with their acidic or basic properties determined by the balance between the inherent Lewis acidity of the cation and the inherent Lewis basicity of the attached groups, either H2O, OH-, O2-, or some combination of these. In the examples considered to this point, Mz+ has been a metal cation; however, it need not be. Mz+ can be any cation of interest. For example, one can imagine the compound CH3CCl3 (1,1,1-trichloroethane) as composed of chloride ions and the cation, CH3C3+. Assuming a maximum coordination number of four for carbon, sequence 6 begins with the hydrated cation, CH3C(H2O)33+, species A. Three succesive deprotonations lead to the neutral hydroxide, CH3C(OH)3, which then loses one molecule of water to form acetic acid, CH3COOH. Similarly, the Bronsted acid, HX (X = Cl, Br, I, NO3, ClO4, and so on), is a Lewis adduct of the cation H+ and the base, X-. On dissolution in water, the cation proceeds to point A in sequence 6 to form H(H2O)+. Although this is a strong acid, there is no possibility of further progress along the sequence (i.e., proton transfer to water simply regenerates H3O+). Thus all typical Bronsted acids are compatible with the ideas based on sequence 6.
The Question of Reality. There are those who would object to the conceptual framework discussed in the paragraphs above on the grounds that cations such as Cl7+, S6+, and CH3C3+ have no objective reality; that is, they have not been detected as species with independent existence. In contrast, less highly charged cations such as Na+, Mg2+, and Al3+ are "real" chemical species. If we ignore the fact that we consider Cl7+ and S6+ real enough to assign radii to them (6,16), I would not dispute this distinction. However, I argue that it is not relevant to the conceptual utility of scheme 6, which is intended only as a way of thinking about the interaction of a cation with electron pairs. Similar situations obtain with the concepts of oxidation state and resonance, both of which are extremely useful as ways of thinking, but neither of which has any objective reality. Scheme 6 is a unifying conceptual framework within which the acid/base properties of seemingly very different molecules may be viewed in common terms.
Some Applications. Application of the ideas presented above to the observable behavior of several common substances illustrates their utility.
First, the categorization of element oxides as basic, amphoteric, or acidic is readily understood. The oxides of 1+ and 2+ cations are adducts of very weakly acidic cations (e.g., Na+ or Mg2+) and the powerfully basic oxide anion. The behavior of the oxide is dominated by the basicity of O2-, which is barely reduced by the weakly acidic cation, so the compound is a basic oxide. In contrast, even three O2- ions are insufficient to offset the powerful acidity of S6+ in SO3. In this case, the oxide has exposed acidity: it is an acidic oxide. The oxide of a 3+ or 4+ cation is essentially neutral; that is, the cation acidity is balanced by the basicity of O2-. Such oxides are amphoteric because they may respond to either acids (via the O2- ion) or bases (via the 3+ or 4+ cation). In general, oxides of 1+ and 2+ cations are basic; of 3+ (and some 4+) cations, amphoteric; and of cations > 4+, acidic.
NaOH and KOH are packaged in pellets, which rapidly become wet when exposed to air. If left standing in the air, the pellets pick up enough atmospheric water to dissolve themselves; they are deliquescent. A traditional introductory explanation of this phenomenon might be that NaOH and KOH are "hygroscopic" (i.e., have an "affinity" for water). Of course, this is descriptive, not explanatory. In terms of the framework above, NaOH is viewed as an adduct of Na+, a cation with minimal inherent acidity, and OH-, a strong base. Na+ is unable to neutralize the basicity of OH-, so NaOH has "exposed basicity." To neutralize this, it spontaneously absorbs water from the air, which interacts via a positively charged hydrogen atom with the lone pairs of OH-.
AlCl3 is a white powder that comes packed in tightly sealed bottles. With repeated opening of the bottle, even for brief time periods, the powder becomes sticky with moisture. Prolonged exposure again results in spontaneous dissolution. In this case, Al3+ has moderate acidity that is not offset by Cl-, which has almost no tendency to donate an electron pair; i.e., there is exposed acidity. Thus AlCl3 absorbs water from the air to offset the acidity of Al3+, eventually forming Al(H2O)63+.
A great variety of salts occur or are sold as hydrates. The water of hydration neutralizes residual acidity or basicity of the anhydrous salt. In some cases, the cation necessitates its presence; in other cases the anion. For example,
Generally speaking, the carbonates, hydroxides, and sulfides of cations with z > 2+ are insoluble. In these cases, cation and anion are more effectively neutralized by each other in the "adduct" than either would be by water, and the salt fails to dissolve. On the other hand, the carbonates, hydroxides, and sulfides of group 1 cations are soluble. The cation-anion adducts have residual basicity that can be offset by dissolution in water.
Finally, NaCl is a white crystalline solid that has very little tendency to absorb water from the air; it stays dry, even when left exposed (except on hot, humid days!). NaCl is an adduct of non-acidic Na+ and non-basic Cl-. There is no exposed acidity or basicity, hence no absorption of atmospheric water.
Conclusions. I have attempted to show that the Bronsted acidity/basicity of species such as Ca(H2O)62+, H3PO4, CH3COOH, and HCl can be understood conceptually in terms of the Lewis acidity of the "parent" cation (Ca2+, P5+, CH3C3+, and H+ in the species cited). The cation is imagined to proceed sequentially, via proton loss, through the series of Lewis adducts in scheme 6 until a species capable of existence in water is formed. The inherent acidity of the parent cation is neutralized to a greater or lesser extent in the adduct by attached basic species that have their genesis in the water molecule. Residual acidity or basicity in the adduct is the source of its subsequent acid/base reactivity.
Acknowledgments. I would like to thank Russell S. Drago for schooling me in Lewis acid-base ideas and their sweeping applications.
References