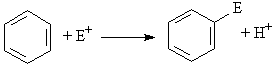
In electrophilic aromatic substitution, the benzene ring acts as an electron donor (Lewis base or nucleophile, ArH) and reacts with an electron acceptor (Lewis acid or electrophile, E+). The result of electrophilic aromatic substitution reactions is that an aromatic hydrogen (ArH) is replaced by some other atom or functional group (ArE). ArH + E+ ---> Ar-E + H+ or
Like an alkene, the double bonds of benzene are nucleophilic and will react with electrophiles (e.g., X2, NO2+, SO3, RX+, and RCO+). But unlike alkene reactions, the outcome is a substitution and not an addition.
| Reaction |
|
|
Product |
|
|
|
||
| Halogenation |
|
|
|
|
|
|
|
|
| Nitration |
|
|
|
| Sulfonation |
|
|
|
| Alkylation |
|
|
|
| Acylation |
|
|
|
A1. Halogenation: Bromination, Chlorination, and Iodination
A1a. Bromination: ArH + Br2 ----> Ar-Br
The electrophile in bromination is simplistically stated
to be Br+. A better description is that the electrophile is
a polarized bromine molecule, FeBr4-Br+,
generated by the catalyst FeBr3.
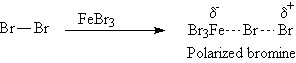
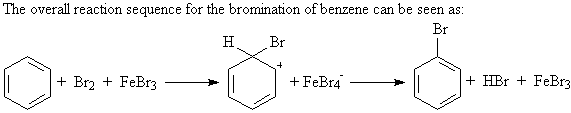
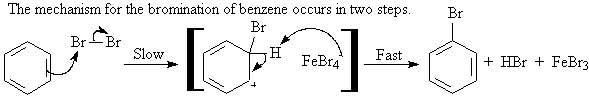
A1b. Chlorination and Iodination
Both chlorination and iodination occur by mechanistic
pathways similar to bromination. The catalyst for chlorination is FeCl3.
Iodine is unreactive and requires an oxidizing agent such as CuCl2
for the generation of the electrophile, I+.
I2 + 2 Cu2+ ----> 2 I+ + 2 Cu1+
A2. Nitration: ArH + HNO3 ----> Ar-NO2
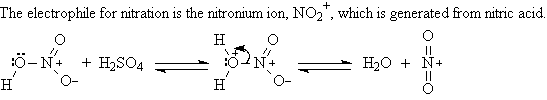
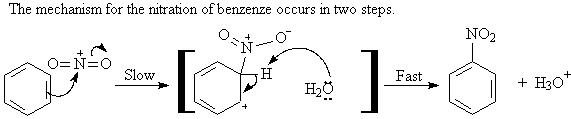
Aryl nitro groups can be reduced to an amino group.
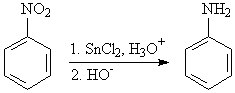
A3. Sulfonation: ArH + SO3 ----> Ar-SO3H
Fuming sulfuric acid, a mixture of H2SO4
and SO3, is used to sulfonate a ring. The product of the sulfonation
is a benzenesulfonic acid,
Ar-SO3H.
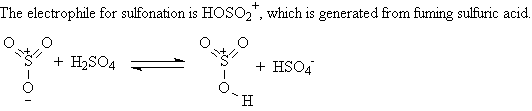
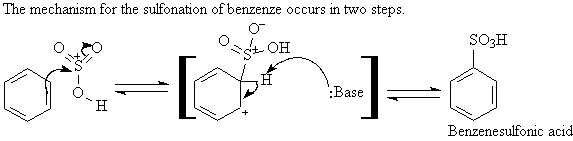
Sulfonation reactions are reversible.
Aromatic sulfonic acids can be further treated to produce phenols. The process, known as alkali fusion, is however limited to alkyl substituted aromatic sulfonic acids.

A4. Alkylation: ArH + R-X ----> Ar-R
Alkylation of the ring is known as the Friedel-Crafts
reaction. The electrophile is a carbocation created from an alkyl halide
in the presence of AlCl3.
Note: At this point it should be obvious that all these reactions have the same mechanism; we just need to know the conditions (i.e., nucleophile, electrophile).
There are four limitations to Friedel-Crafts reactions:
a. Only alkyl halides can be used
(i.e., not vinyl or aryl halides).
b. The reaction is not successful
with a ring substituted with amino groups (-NH2) or with electron-withdrawing
groups
such as nitro, cyano, acids or esters. (It will be shown later that such
rings are any substituted aromatic
rings
less reactive than a halobenzene.)
c. It is difficult to stop at monosubstitution.
This can be limited by using an excess of the hydrocarbon and running
the reaction
at lower temps. When an excess of the hydrocarbon is used, the electrophile
is more likely to encounter
an unsubstituted
ring than the substituted ring.
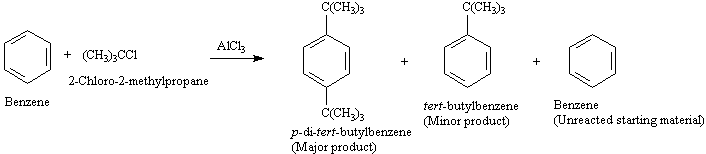
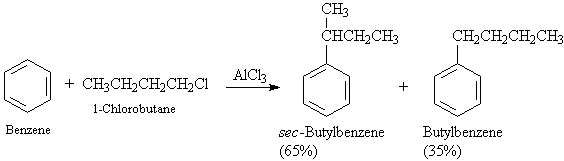
d. Since a carbocation is involved,
rearrangements may occur, particularly if primary alkyl halides are involved.
These carbocation
rearrangements can occur either by hydride shifts or alkyl shifts. Lower
temps tend to keep
down the rearrangement
of the carbocation.
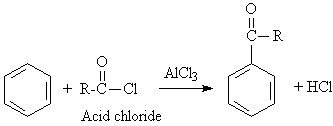
A5. Acylation: ArH + RCOCl ----> Ar-COR
In the Friedel-Crafts acylation, an acid chloride is
used to introduce an acyl group to the ring. This type reaction is a way
to get around the problem of rearrangement and polysubstitution found in
Friedel-Crafts alkylations. It will be shown later that the carbonyl group
can be reduced to an alkyl group.
B. Substituent effects
Substituents affect both the reactivity of the ring and
the orientation of the product.
Some substituents make the ring more reactive than benzene, while others make the ring less reactive than benzene. In the case of nitration, a hydroxyl group, -OH, makes the ring 1000 times more reactive than benzene, while a nitro group, -NO2, makes the ring more than 10 million times less reactive.
Either the ortho, para products or the meta product will be favored depending on upon the substituent present on the ring. If the cation is stabilized, the ortho, para products predominate and if the cation is destabilized then the meta product predominates. If the first group withdraws electrons from the cation, it destabilizes the cation and if it donates electrons it stabilizes it.
Substituents can be classified into three groups:
1. Ortho, para-directing activators.
Functional groups in this category include R (alkyl), -NH2,
-NHR, -NR2 (amino),
-NHCOR (amide),
-OH (hydroxyl), and -OR (ether).
2. Ortho, para-directing deactivators. The halogens (F, Cl, Br, and I) are the most important substituents in this group.
3. Meta-directing deactivators. Functional
groups in this category include -NO2 (nitro), -SO3H
(sulfonic acid),
and all carbonyl
compounds: -CO2H (acids), -CO2R (esters), -COH (aldehydes),
and COR (ketones).
There are no meta-directing activators.
Reactivity and orientation are best explained by inductive effects and resonance effects.
The inductive effect is the withdrawal or donation of electrons through a sigma bond (s bond) due to the electronegativity and the polarity of the bonds in the functional groups. Groups bonded to the ring which are more electronegative will withdraw electrons. Alkyl groups will inductively donate electrons to the ring.
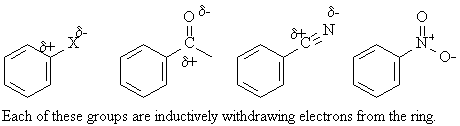
The resonance effect is the withdrawal or donation of electrons through a pi bond (p bond) due to the overlap of a p orbital on the substituent with a p orbital on the ring by resonance.

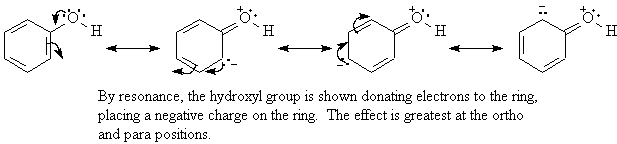
The common feature of activating groups is that they donate electrons to the ring. In most activating groups, the atom of these groups which is directly bonded to the ring will have lone pairs of electrons. Electron-donating groups help to stabilize the carbocation intermediate from electrophilic addition and cause it to form faster. Alkyl groups have an electron-donating inductive effect.
The common feature of deactivating groups is that they withdraw electrons from the ring. In most deactivating groups, the atom of these groups which is directly bonded to the ring will have multiple bonds or will have a positive formal charge. The groups are electron-withdrawing groups. Electron-withdrawing groups destabilize the carbocation intermediate from electrophilic addition and cause it to form slower.
With the halogens the two effects (inductive vs. resonance)
are closely balanced and the effect is small and deactivating but the directing
effect is ortho- and para-. The electron-withdrawing inductive effect is
more important to the overall cation stability, but the resonance electron-donating
effect dominates the cations resulting in ortho- and para- substitution.
The electron-withdrawing inductive effect is slightly greater than the
electron-donating resonance effect so the halogens are deactivating. Though
weak, the electron-donating resonance effect if felt only at the ortho-
and para- positions.
|
(Electron-donating) -OH (hydroxyl) -OR (ether) -NHCOR (amide) -R (alkyl) -Ar (aryl) |
(Electron-withdrawing) -NO2 (nitro) -CO2H (acid) -CN (cyano) -CO2R (ester) -COR (ketone) -CHO (aldehyde) |
C. Trisubstituted Benzenes: Additivity of Effects
Rule 1. If the groups reinforce, there
is no problem.
Rule 2. If they conflict the more
activating group wins.
Rule 3. It is hard to get a new group
between two existing groups; it'll go into the more open slot for steric
reasons.
See questions at http://www.cem.msu.edu/~parrill/ch16/index.html
D. Miscellaneous Reactions
D1. Nucleophilic Aromatic Substitution
In electrophilic aromatic substitution, the benzene ring
acts as an electron donor (Lewis base or nucleophile) and reacts with an
electron acceptor (Lewis acid or electrophile).
The result of electrophilic aromatic substitution reactions is that an aromatic hydrogen is replaced by some other atom or functional group.
In nucleophilic aromatic substitution, halogens are substituted by a nucleophile only if the aryl halides have electron-withdrawing groups in a position ortho or para to the halogen.
The result of nucleophilic aromatic substitution reactions
is that a halogen is replaced by some other atom or functional group.
Electrophilic substitutions are favored by electron-donating
groups which stabilize the carbocation intermediate.
Nucleophilic substitutions are favored by electron-withdrawing
groups which stabilize the carbanion intermediate.
Electron-withdrawing groups that deactivate a ring for
EAS, activate a ring for NAS.
These groups are meta-directors in EAS but are ortho-
para-directors in NAS.
Cannot occur by SN2 because the ring is in
the way on the backside. Cannot occur by SN1 because the vinyl
cation is unstable. So undergoes addition to form the anion. Since this
is very unstable, it only occurs if there are electron withdrawing groups
o, p to stabilize the anion.
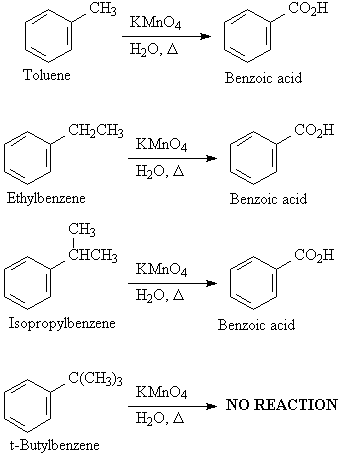
D2. Side Chain Oxidation
Alkyl side chains are oxidized down to the carboxylic
acid if the chain has a benzylic hydrogen.
[O]
Ar-R -----> ArCO2H
[O] = KMnO4, HNO3, or K2Cr2O7
D3. Bromination of Alkylbenzene Side Chains
Just as the allylic position of an alkene is brominated
by N-bromosuccinamide (NBS) so the benzylic position of an alkylbenzene
side chain is susceptible to bromination by NBS. The reagents include NBS
in the presence of benzoylperoxide (PhCO2)2, and
carbon tetrachloride as a solvent.
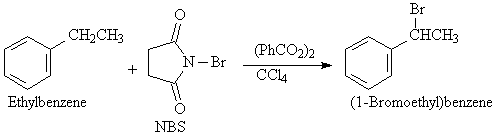
D4. Catalytic Hydrogenation
D4a. Selective hydrogenation of alkene double bond side
chains can be accomplished by catalytic hydrogenation using hydrogen gas
at atmospheric pressure and a palladium catalyst.
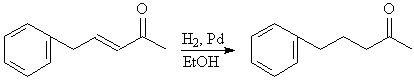
D4b. Hydrogenation of the ring requires a platinum catalyst at several hundred atmospheres of pressure.
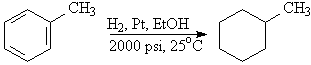
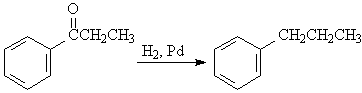
D5. Side chains with carbonyls (including those formed when acylating the ring in the Friedel-Craft) can be reduced to an alkyl group.
E. Synthesis of Trisubstituted Benzenes
The order of addition is dictated by the directing effects
of the substituents.
To prepare p-nitrotoluene starting
with benzene. 1. Alkylate 2. Nitrate
To prepare p-bromoacetophenone starting
with benzene. 1. Brominate 2. Acylate
To prepare m-chloroethylbenzene starting
with benzene. 1. Acylate 2. Chlorinate 3. Reduce
See others at http://www.cem.msu.edu/~parrill/ch16/index.html
Suggested chapter 16 problems:
24, 27, 28, 29, 30, 31, 32, 33, 35, 40, 47, 48, 49, 50,
70